Es freute uns sehr, zu unserem zehnjährigen Bestehen auch im medizinischen Teil unseres Jubiläumskongresses einige neue und interessante Entwicklungen präsentieren zu können. Dafür danken wir allen unseren Referenten. Im folgenden ein Update der wichtigsten Neuerungen zum Stand der medizinischen Forschung.

Den Samstag nutzten wir, unsere Familien, Ärzte und Therapeuten auf den neusten Stand der Forschung an Gangliosidosen zu bringen. Alle Fotos: Patty Varasano
Unser medizinischer Beirat, Dr. Eugen Mengel, Gründer und CEO der SphinCS, eröffnete den medizinischen Teil unserer Jubiläums-Tagung. Und er hatte einige spannende Neuigkeiten dabei. Zu gleich drei Behandlungsansätze – der Substratreduktion, dem Chaparone-Effekt und der Enzymersatztherapie gebe es neue Entwicklungen. Daneben fasziniere ihn eine spannende Forschungsarbeit Freiburger Wissenschaftler.
Doch der Reihe nach: Weltweit startet die Pharmafirma „Azafaros“ in diesen Wochen eine klinische Studie, um den von ihnen entwickelten Wirkstoff Nizubaglustat für GM1, GM2 und Niemann-Pick Typ C als Arzneimittel zuzulassen. Es handele sich um eine 18-monatige doppelblinde, randomisierte, placebokontrollierte, multizentrische Phase-3-Studie zur Bewertung der Sicherheit und Wirksamkeit von Nizubaglustat (AZ-3102) bei spätinfantilen und juvenilen Formen der GM1-Gangliosidose oder GM2-Gangliosidose sowie bei spätinfantilen und juvenilen Formen der Niemann-Pick-Krankheit Typ C. Nizubaglustat ist eine Substanz die man einmal am Tag oral einnimmt. Das Potenzial für klinische Wirkungen bei GM1/GM2-Gangliosidose wird durch ein nichtklinisches Modell der Sandhoff-Krankheit gestützt, bei dem Nizubaglustat die Überlebenszeit verlängerte und die Leistungsfähigkeit in Verhaltensendpunkten verbesserte.
Wer kommt für die Studie infrage?
Spät-infantile und juvenile GM1 und GM2 Patienten zwischen 4 und 20 Jahren, erste neurologische Symptome müssen zwischen dem ersten und dem zehnten Lebensjahr aufgetreten sein. Eine SARA-Testung müsse möglich sein und es dürften keine ganz schweren Schluckstörungen vorliegen. Die SphinCS in Hochheim ist das einzige Institut, das in Deutschland als Prüfstelle für die Studie eingereicht wurde. In der Schweiz ist die Klinik für Neurologie und Neuropädiatrie des Universitätsspital Bern Prüfstelle für die Studie.

Unser medizinischer Beirat, Dr. Eugen Mengel, hatte viele Neuigkeiten zum Stand der Forschung bei GM1- und GM2-Gangliosidosen.
Ganz anders wirkt Aloxistatin. Es gehört im weitesten Sinne zu den Chaperonen. Die sogenannten Chaperone hängen sich an fehlerhaft gefaltete Enzyme und stabilisieren die Struktur des Enzyms. Damit wird der Abbau des Substrates im Lysosom wieder möglich. Aloxistatin sei aber kein echter Chaperon-Effekt, so Mengel. Es ist ein „Enzyme Enhancement“, aber aus einem anderem Grund wie bei einem Chaperon. Dennoch könne Aloxistatin die Aktivität von ß-Gal oder Hex A steigern, verbessern oder sogar wiederherstellen.
Dabei sei Aloxistatin eine alt bekannte Substanz – nebenwirkungsarm und sicher. D-Orphan, eine kleine pharmazeutische Firma aus der Schweiz, habe es auf der Suche nach MPS-Medikamenten zufällig wieder entdeckt. Ursprünglich habe man es nur für GM1 im Blick gehabt, weil eine häufige Mutation bei GM1 typisch für die Fehl-Fältelung der Enzyme ist. Doch auch bei GM2 gebe es Mutationen, die eine Fehl-Fältelung des Enzyms verursachen würden, so Mengel. Mindestens eine solche Mutation müsse ein Patient haben, damit Aloxistation wirken könne.
Um dies belegen zu können, habe Dr. Mengel ein Studienprotokoll erstellt, das vom Bundesinstitut für Arzneimittelsicherheit (BfArm) und der Ethikkommission genehmigt werden müsse. Die Anhörung beim BfArm sei noch in diesem Jahr terminiert. Wenn alles gutgehe, müsse freilich die Substanz noch besorgt werden.
Gute Nachrichten gebe es von der japanischen Pharmafirma JCR, die an einer Methode arbeiten, die Enzymersatztherapie über die Blut-Hirn-Schranke zu bekommen. Bei der Enzymersatztherapie wird das für den Abbau der im Lysosom notwendige Enzym von außen eingebracht. Bei unseren Krankheitsbildern (GM1 und GM2) scheiterte das bislang an der Blut-Hirn-Schranke. Die Forschung könne nun weitergehen, nachdem die Pharmafirma einen Investor gefunden habe, und deshalb das Enzym für die Grundlagenforschung in großem Maßstab produzieren kann.
Zudem berichtete Dr. Eugen Mengel von einem überraschenden Forschungsansatz eines Freiburger Foscherteams, die sich den Ablauf der GM2-Erkrankung genau angesehen und dabei eine Art Teufelskreis aus Entzündung und neuronaler Schädigung und dessen Ursache definiert haben. Die Forschenden haben darüber hinaus gezeigt, dass man diesen Teufelskreis durchbrechen kann, wenn man die Mikroglia, die das Lysosomenenzym zu den Neuronen liefert, mit anderen, peripher stammenden mikroglia-ähnlichen Zellen ersetzt. Mit diesen „Neuen“ lässt sich das Gleichgewicht im Gehirn wiederherstellen, und neurodegenerative Schäden können zurückgedreht werden. Es sei extrem spannend, wie diese Forschungen weitergehen werden, so Mengel.
Stand N-Acetyl-L-Leucine und Ataxie

PD Dr. Tatiana Brémová-Ertl war 2017 erstmalig als Referentin bei unserer Familienkonferenz zu Gast.
Ein weiteres Forschungs-Update bekamen wir von Priv.-Doz. Dr. med. Tatiana Brémová-Ertl, von der Klinik für Neurologie und Neuropädiatrie, Universitätsspital Bern, Schweiz. Brémová-Ertl ist stellvertretende Leiterin des dortigen Zentrum für Seltene Krankheiten und Leiterin der Sprechstunde für Neurometabolik.
Zunächst widmete sich Ihr Vortrag dem Thema Ataxie, die gerade bei unseren jugendlichen und erwachsenen Patientinnen für Einschränkungen sorgt. Der Begriff Ataxie komme aus dem Griechischen und bedeutet Unordnung, Unregelmäßigkeit. Der Begriff umfasse Störungen der Bewegungskoordination, des Ablaufs, der Feinsteuerung einzelner Bewegungen und des Zusammenspiels von komplexen Bewegungsabläufen. Brémová-Ertl erklärte die unterschiedlichen Formen der Ataxie, wie sie entstehen und zu welchen Symptomen sie führen. Zudem erläuterte sie die Entwicklung von Scores, wie zum Beispiel den SARA-Score, mit dessen Hilfe die Wirksamkeit von Medikamenten anhand der Ataxie gemessen werden können. SARA steht für „Scale for Assessment and Rating of Ataxia“.
Im Anschluss gab sie uns ein Update zur Situation des Wirkstoffes N-Acetyl-L-Leucine (NALL). Tatiana Brémová-Ertl war in den Entwicklungsprozess diese Wirkstoffes damals noch am deutschen Schwindelzentrum des Universitätsklinik München eng eingebunden. Unsere Selbsthilfegruppe beteiligte sich von Anfang an an den individuellen Heilversuche mit einem in Frankreich zugelassenen Medikament gegen Schwindel und Migräne.
Erstmalig sprach Tatiana Brémová-Ertl 2017 auf einer unserer Konferenzen über das Thema. Zunächst ging es darum, die Wirksamkeit für Niemann Pick Typ C und auf unser Betreiben hin auch für Tay-Sachs und Sandhoff in individuellen Heilversuche zu belegen, um den Wirkstoff dann zu optimieren und weiterzuentwickeln. So wurde aus N-Acetyl-DL-Leucine das heute verwendete N-Acetyl-L-Leucine (NALL).
N-Acetylierung kreiere ein Prämolekül, das sehr effektiv in die Zellen aufgenommen werde, so Brémová-Ertl. Die hohe L-Leucin Konzentration in den Zellen sei therapeutisch und verbessere den Energiestoffwechsel der Zellen. N-Acetyl-L-Leucine wirke neuroprotektiv und verhindere den Nervenverlust. Für Niemann-Pick Typ C ist das Medikament in den USA bereits zugelassen. Die Pharma-Firma Intrabio hat einen Antrag für die Anerkennung von NALL in GM2 Indikation gestellt. Dies wurde abgelehnt, jedoch mit einer Möglichkeiten “PatientInnen-Wege” aufzuzeichnen, um die erweiterte Zulassung für GM2 zu erhalten.
Grundlagenforschung und Mausmodell zur Gentherapie
Zum ersten Mal sprach Professor Dr. Wolfgang Baumgärtner bei einer unserer Konferenzen. Prof. Baumgärtner forscht an der Tierärztlichen Hochschule Hannover. Dort war er über viele Jahre Leiter des Instituts für Pathologie und Leiter der Abteilung Diagnostik und Leiter der Forschungseinheit: Neuropathologie und Neuroimmunologie. 2006 bis 2009 erforschte er im Rahmen eines DFG-Forschungsstipediums die Charakterisierung des molekularen Defekts von GM1-Gangliosidose bei Alaskan Huskies. In einem weiteren DFG-Projekt ging es um die Korrektur eines genetischen Defekts der GM1-Gangliosidose bei Alaskan Huskies mittels Zinkfinger-Nukleasen.

Prof. Wolfgang Baumgärtner betreibt in Hannover Grundlagenforschung an GM1.
Baumgärtner gab uns einen spannenden Einblick in seine Grundlagenforschung: Natürlich vorkommende genetische Defekte im Glb1-Gen gebe es nicht nur beim Menschen, sondern auch bei Hunden, Katzen, Kühen, Schafen, amerikanischen Schwarzbären und anderen Tieren. Künstlich induziert (Knock out) in verschiedenen Mausmodellen. Klinische, morphologische und biochemische Merkmale erkrankter Alaskan Huskies ähnelten Erkrankungen der späten infantilen/juvenilen Form (Typ 2) der humanen GM1-Gangliosidose. Knock-Out-Mäuse hingegen ahmen hinsichtlich histologischer und immunhistochemischer Befunde die adulte Form der GM1-Gangliosidose beim Menschen nach.
Sein aktuelles Projekt ist eine „Neuartige Gentherapie-Strategie zur Korrektur des GM1-Gangliosids in einem Mausmodell unter besonderer Berücksichtigung von frühen und späten Phasen der Erkrankung“.
Baumgärtner berichtete auch von seinen präklinischen Studien an dem Substrathemmer Sinbaglustat. Die Langzeitbehandlung von Glb1-Knock-out-Mäusen mit Sinbaglustat führte durch die reduzierte neuronale Akkumulation von GM1 zu einem verzögerten Beginn und einem verlangsamten Fortschreiten der klinischen Erkrankung. Allerdings konnte eine neuronale Akkumulation von GM1 nur bedingt vermindert werden.
Der therapeutische Ansatz sei die Hemmung von an der Gangliosidose beteiligten Enzymen durch den Iminozucker (Azazucker) Sinbaglustat. Der sei ein starker Hemmer der extralysosomalen Glucosylceramidase beta 2 (GBA2, Glucocerebrosidase) und ein schwacher Hemmer der Glucosylceramid-Synthase (GCS). Leider werde Sinbaglustat, das auch bei GM2 Anwendung finden könnte, von der Schweizer Pharmafirma Idorsia aus Kostengründen aktuell nicht weiter verfolgt.
Unsere Studien „8-in-1“ und PRADO

Dr. Hannah Arnold war auch zum ersten Mal bei unserer Familienkonferenz zu Gast.
Dr. med. Hannah Arnold gab einen ersten Überblick über die seit März 2025 laufende Prado-Stduie (Projekt Adult Onset). Ziel der PRADO-Studie ist eine systematische Erfassung der psychischen Symptome bzw. psychiatrischen Komorbiditäten von Patienten mit M. Tay-Sachs und Morbus Sandhoff. Dabei gehe es insbesondere auch um die Rolle des Kleinhirns. Denn Beeinträchtigungen der Kleinhirnfunktion werden mit der Entwicklung von akuten Psychosen in Verbindung gebracht. GM2-Gangliosidosen könnten als Modellerkrankung für andere Erkrankungen, die mit einer Störung der Kleinhirnfunktion einhergehen, dienen.
Kriterien zum Studieneinschluss:
- juvenile oder adulte Verlaufsform einer GM2-Gangliosidose
- Alter von mind. 12 Jahren
In die Studie konnten insgesamt 17 Probanden eingeschlossen werden:
- 3 Probanden mit M. Sandhoff
- 14 Probanden mit einer Tay-Sachs-Erkrankung
- Alter: 16 Jahre bis 61 Jahre
- 6 Probanden mit einer juvenilen und 11 Probanden mit einer adulten Verlaufsform
Es wurden kognitive Tests und ein CCAS-Test durchfgeführt, der die Kleinhirnfunktion prüft. Zudem wurden in psychiatrischen Kurzinterviews die wichtigsten psychiatrischen Symptome abgefragt, und zwar sowohl aktuelle als auch Symptome in der Vergangenheit.
Erste Ergebnise:
- Bei den untersuchten Patienten mit GM2-Gangliosidosen bestanden keine relevanten kognitiven Defizite, Hinweise auf eine Kleinhirnschädigung bei allen Probanden
- An psychiatrischen Diagnosen konnten am häufigsten Angststörungen, Depressionen und Psychosen gefunden werden
Dr Eugen Mengel gab ein Update der „Acht-in-Eins-Studie“, die unsere Selbsthilfegruppe zusammen mit der SphinCS vor fünf Jahren ins Leben gerufen hat. Es ist eine Register- und natürliche Verlaufsstudie für alle GM1- und GM2-Gangliosidosen. Mittlerweile wurden 84 Patientinnen und Patienten aufgenommen. Von den acht Gangliosidosen sind sieben vertreten:
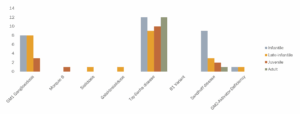
Eugen Mengels Fazit nach fünf Jahren „8-in-1“-Registerstudie:
- Die Daten würden die Notwendigkeit wirksamer Therapien untermauern.
- Wichtig sei eine frühzeitige Diagnose , dazu müsse man auf die Eltern hören, aber auch ein Neugeborenenscreening sei hierzu wichtig.
- Unglaubliche Zahl von 84 Patienten
- Prado helfe, psychische Erkrankung besser zu verstehen
- Patienten mit Late-Onset GM2: Haben eine Erkrankung der Nerven, die Muskeln versorgen und eine Kleinhirnerkrankung
- Medikamentenentwickler nehmen uns war
Hinterlasse einen Kommentar